

Retinoblastoma is a rapidly developing cancer that develops from the immature cells of a retina, the light-detecting tissue of the eye. It is the most frequently occurring neoplasm of the eye in childhood, occurring in about 1 in 14,000–18,000 live births. Thus, an estimated 8000 cases will occur worldwide. A higher incidence is noted in developing countries.
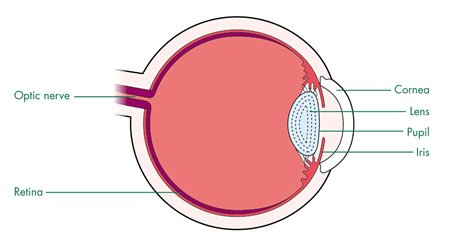
There are two distinct clinical forms of retinoblastoma.
- The first is a hereditary form (40% of cases), characterized by the presence of germline mutations of the RB1 gene. Hereditary retinoblastoma may be inherited from an affected survivor (25% cases) or may be the result of a new germline mutation (75% cases).Usually both eyes are affected ( bilateral) but sometimes it occurs only in one eye.
- The second is a non-hereditary form (60% of cases ). In this type, in 90% of cases, there is involvement of only one eye
Causes of Retinoblastoma
In children with the heritable genetic form of retinoblastoma there is a mutation on chromosome 13, called the RB1 gene. The genetic codes found in chromosomes control the way in which cells grow and develop within the body. If a portion of the code is missing or altered (mutation) a cancer may develop.
The defective RB1 gene can be inherited from either parent.In some children, however, the mutation occurs in the early stages of fetal development.
Inherited forms of retinoblastomas are more likely to be bilateral. In addition, they may be associated with pinealoblastoma (also known as trilateral retinoblastoma) with a dismal outcome.
In case of non-heritable retinoblastoma, the cause is unknown
Clinical Manifestations
Almost 80% of children with retinoblastoma are diagnosed before 3 years of age and diagnosis in children above 6 years of age is extremely rare. Patients with bilateral retinoblastoma tend to present at a younger age (usually before 1 year of age) than do children with unilateral disease, who often present in the second or third year of life.
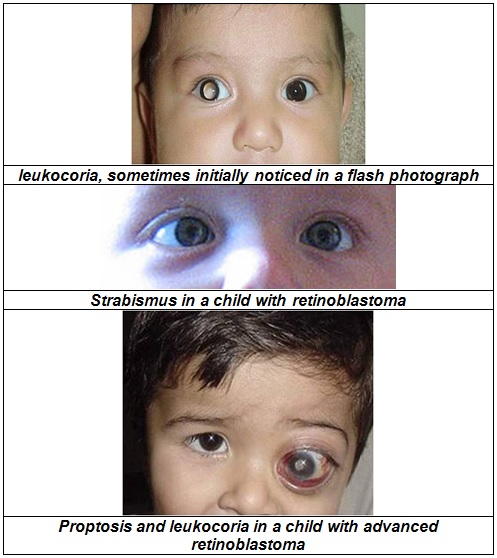
The most common and obvious sign of retinoblastoma is an abnormal appearance of the pupil. Leukocoria, also known as amaurotic cat’s eye reflex, denotes a white appearance of the pupil . Some children with retinoblastoma can present with a squint, commonly referred to as “cross-eyed” or “wall-eyed” (strabismua). Other signs and symptoms include deterioration of vision, a red and irritated eye with glaucoma and faltering growth or delayed development.
Retinoblastoma can present with advanced disease in developing countries and in this case,eye swelling or proptosis is a common finding.
Diagnosis
Screening for retinoblastoma should be part of a “well baby” screening for newborns during the first three months of life. In familial cases, the diagnosis is usually made through screening, Children with a family history of retinoblastoma should receive a thorough ophthalmological examination under anesthesia shortly after birth and periodically thereafter.
The diagnosis of retinoblastoma is usually established by an experienced ophthalmologist. The retina should be examined under anesthesia. Retinoblastomas can be diagnosed just by their appearance. So taking a tissue sample or biopsy is not necessary
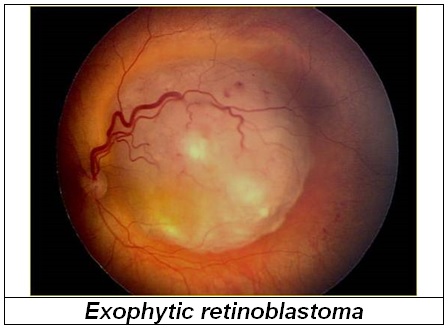
If a diagnosis of retinoblastoma has been made, the child will have to undergo several test to determine the size and extent of the tumour and whether it has spread to the surrounding tissues or distant sites.This is called staging
These test may include –
- Imaging studies, such as computerized tomography (CT), magnetic resonance imaging (MRI), and ultrasound. Ultrasound can help define the height and thickness of the tumor.
- Bone marrow examination to determine any spread of the cancer to the bone marrow
- Lumbar puncture where a sample of the fluid around the brain.and spinal cord is taken and examined under the microscope for any cancer cells
- Bone scan – this involves taking X-ray pictures to look for spread to the bones
- Blood test. This may be taken for genetic testing for the Rb gene. The results of this test can take some months.
Treatment of Retinoblastoma
This depends on the number, site and size of the tumours in the eye and on whether the tumour has spread to other tissues. The aim of treatment is to get rid of the cancer and secondly to try to preserve vision.
Thus treatment of retinoblastoma can nvolve 2 different scenarios: (i) eye-preserving treatment, when intraocular disease allows for a conservative approach (i.e., to save the patient’s vision), and (ii) life-saving treatment, when eye conservation is not possible, and treatment is aimed at saving the patient’s life.
The various treatment modalities for retinoblastoma includes :-
Focal therapy
- Focal treatments, are used for small tumors (<3–6 mm) usually in patients with bilateral disease, and in combination with chemotherapy.
- Laser photocoagulation- Laser photocoagulation is recommended only for small posterior tumors. A laser is used to heat the tumour. Two or three monthly sessions are usually required.
- Cryotherapy- Cryotherapy freezes the tumour. Cryotherapy may be used as primary therapy for small peripheral tumors or for small recurrent tumors previously treated with other methods.
- Thermotherapy- Thermotherapy involves the application of heat directly to the tumor, usually in the form of infrared radiation. It is also used for small tumors.
- ChemotherapyChemotherapy or the administration of anti cáncer drugs was previously used only to treat metastatic disease; However, since the mid-90s, it has been widely used as the primary treatment for intraocular disease not amenable to local therapy. Systemic chemotherapy is used to decrease the tumor size and make the tumors suitable for local therapy. This approach, called chemoreduction, aims to prevent enucleation or the use of external beam radiotherapy in selected cases.
- EnucleationWhen the tumour is very large and visión is lost, the child will have to undergo an operation to remove the eye An orbital implant or prosthesisis is then fitted . Most patients with unilateral disease present with advanced intraocular disease and therefore usually undergo enucleation, which results in a cure rate of 95%. In bilateral Rb, enucleation is usually reserved for the worse eye without useful vision or eyes that have failed all known effective therapies .
- External beam radiotherapyRadiotherapy in combination with focal treatments can provide excellent tumor control because retinoblastoma is a very radiosensitive tumor. However, since radiation therapy increases the risk of secondary malignancies in retinoblastoma survivors, contemporary management of intraocular retinoblastoma is designed to avoid or delay its use. Radiotherapy is mainly as salvage method for eyes that have failed to respond to chemotherapy and focal treatments, usually because of the progression of vitreous and subretinal seeding. Radiation therapy continues to play a major role in the treatment.
- Genetic testingIdentifying the RB1 gene mutation that led to a child’s retinoblastoma can be important in the clinical care of the affected individual and in the care of (future) siblings and offspring.Bilaterally affected individuals and 13-15% of unilaterally affected individuals, are expected to show an RB1 mutation in blood. By identifying the RB1 mutation in the affected individual, (future) siblings, children, and other relatives can be tested for the mutation.If they do not carry the mutation, the child’s relatives are not at risk of retinoblastoma and hence need not undergo the trauma and expense of examinations under anaesthetic. For the 85% of unilaterally affected patients who are negative for the RB1 mutations, neither molecular testing nor clinical surveillance of their siblings is required.
| Last Reviewed | : | 23 June 2016 |
| Writer | : | Dr. Yeoh Seoh Leng |
| Accreditor | : | Dr. Eni Juraida bt. Abdul Rahman |





